Clathrate Hydrates: structure and dynamics
Clathrate hydrates are nanoporous crystalline materials made of a network of hydrogen-bonded water molecules (forming host cages) that is stabilized by the presence of foreign (generally hydrophobic) guest molecules. The natural existence of large quantities of hydrocarbon hydrates in deep oceans and permafrost is certainly at the origin of numerous applications in the broad areas of energy and environmen- tal sciences and technologies (e.g. gas storage). At a fundamental level, their nanostructuration confers on these materials specific properties (e.g. their “glass-like” thermal conductivity) for which the host-guest interactions play a key role [ref.
37]. These interactions occur on broad timescale and thus require the use of multi-technique approach (Neutron scattering, Raman, NMR, Molecular Dynamics Simulations) in which neutron scattering brings unvaluable information [ref.
39]. The investigation of dynamical properties of clathrate hydrates, ranging from intramolecular vibrations to Brownian relaxations reveals the underlying factors governing chemical-physics properties specific to these nanoporous systems.
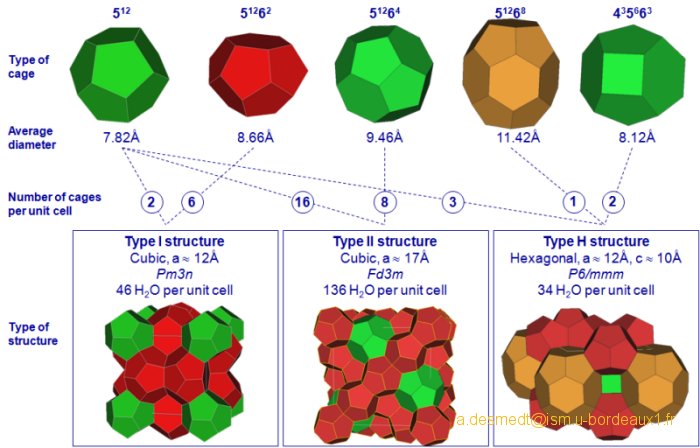
 Structure
Structure
Dynamics
of hydrogen clathrates hydrates
By inserting H2 molecules with the help of a promoter (tetrahydrofuran, noted THF), systems relevant for hydrogen storage application are formed by using relatively soft pressure (of the order of 100 bar) near room temperature. Dynamic properties of hydrogen molecules confined in the small cages of the deuterated THF clathrate hydrate have been investigated at equilibrium by means of incoherent quasi-elastic neutron scattering (QENS). These QENS investigations provide direct experimental evidence about the fundamental aspect of translational diffusive motions of the hydrogen molecules. A comprehensive study of the hydrogen molecules dynamics above 100 K has been achieved through a quantitative analysis of the structure factors (i.e., the spatial extend of the H2 diffusive motion) as well as of the QENS broadening (i.e., the characteristic time of the diffusive motion). On the probed time scale, the H2 molecular translations occur within localized spherical area in the cage with low activation energy of 1.59 ± 0.06 kJ mol–1. The dynamical diameter of H2 molecules varies from 2.08 Å at 250 K to 1.64 Å at 100 K, and the diffusion constant ranges from 0.16 ± 0.03 rad ps–1 at 100 K to 0.49 ± 0.03 rad ps–1 at 250 K. These results indicate that no diffusion between the cages is observed in the picosecond time scale [ref.
40].
Dynamics
and protonic transport in clathrates hydrates
In
the perchloric acid clathrate hydrate HClO4·5.5H2O (type I),
the perchlorate anions are contained inside an aqueous
host crystalline matrix, positively charged because
of the presence of delocalized acidic protons. Our experimental
results demonstrate that the microscopic mechanisms
of proton conductivity in this system are effective
on a time scale ranging from nanosecond to picosecond.
We have studied the relaxation processes occurring on
a nanosecond time scale by combining high-resolution
quasielastic neutron scattering and 1H pulse-field-gradient
nuclear magnetic resonance experiments. The combination
of these two techniques allows us to probe proton dynamics
in both space and time domains. The existence of two
types of proton dynamical processes has been identified.
The slowest one is associated to long-range translational
diffusion of protons between crystallographic oxygen
sites and has been precisely characterized with a self-diffusion
coefficient of 3.5×10-8 cm2/s
at 220 K and an activation energy of 29.2±1.4
kJ/mol. The fastest dynamical process is due to water
molecules' reorientations occurring every 0.7 ns at
220 K with an activation energy of 17.4±1.5 kJ/mol.
This powerful multitechnique approach provides important
information required to understand the microscopic origin
of proton transport in an ionic clathrate hydrate. [ref.
21]
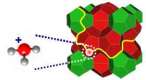
Representation
of the protonic conduction in an acidic hydrate.
Dynamics of methyl-halide clathrates hydrates
CH3I - 17H2O - type II. Methyl
iodide enclosed in the 51264 cage
of cubic II water clathrate has been investigated by
neutron spectroscopy in the temperature regime from
1.8 to 145 K. The three tunnelling bands observed at
the lowest temperature at 497, 372 and 243 µeV
of relative intensities 2:3:5 represent three distinct
adsorption sites of CH3I. Sites of occurrence
probabilities corresponding closely to the observed
line intensities are related to the three types of hydrogen
bonds of the cage. The band at 372 µeV shows fast
spin conversion compared to the two others. The corresponding
site is facing the shortest O–H..O hydrogen bond
with the fastest proton dynamics. Spin conversion may
thus reflect hydrogen jumps within the H-bond network
of host molecules. Jump rates of the order of 10-4
s-1 can be estimated [ref.
17,19]. Above a temperature
T ~ 45 K the methyl dynamics become classical and can
be described by rotational diffusion on a circle. A
second quasielastic process becomes visible at a higher
temperature. Above T ~ 90 K translational freedom
of the CH3I leads to a rattling mode appearing
at ~0.95 meV. A comprehensive study of the dynamic properties of the methyl iodide molecule encapsulated in the large cage of the corresponding type II clathrate hydrates has been performed between 150 K and room temperature. An approach combining quasi-elastic neutron scattering (QENS) and molecular dynamics (MD) simulations provide insights into dynamics across a time scale ranging from tenths to hundreds of picoseconds. MD-derived QENS spectra have been computed with the help of the atomic MD trajectories and directly compared with the experimental ones. The successful agreement between experimental and theoretical spectra allows the validation of the MD simulations, so that a detailed model of dynamic properties is elaborated. The results of both the QENS and MD studies demonstrate that no water molecule relaxation occurs on the probed time scale, whereas relaxation together with a low-energy excitation (observed on experimental and MD-derived QENS spectra) of the guest molecule is observed. Modeling this low-energy excitation shows that the strongly anharmonic rattling of guest molecules within the cage originates from a translational mode (at 1.31 ± 0.03 meV with a damping of 1.49 ± 0.03 meV) coupled to a librational mode (at 0.85 ± 0.05 meV with a damping of 1.19 ± 0.01 meV) of the whole molecule. The localized translational relaxation of the guest molecules within the cage is characterized by a diffusion constant of 1.22 ± 0.06 ps–1 at 270K with low activation energy of 1.5 ± 0.1 kJ/mol. Moreover, whole reorientations of guest molecules are characterized with a diffusion constant of 2.47 ± 0.55 ps–1 at 270 K and a low activation energy of 0.8 ± 0.1 kJ/mol (in addition to fast rotations of the methyl group). By modeling the structure factor and analyzing orientational distribution functions, it is found that the guest molecules are off-centered and undergo strongly anisotropic reorientations within the cages. Guest molecules preferentially adopt orientations that are different from cage to cage due to a static (with respect to the probed time scale) disorder of the H-bond network. The various local environment leads to a distribution of energy landscapes explored by the guest molecules in terms of geometrical characteristic but not in terms of characteristic times of guest molecules dynamics in the present combined MD-QENS investigation. [ref.
38]
CH3Br - 5.75H2O - type I. Methyl
rotational tunnelling, quasielastic scattering and phonon
spectroscopy with neutrons at energies from 40 µeV
to 200 meV is used to characterize the dynamics of CH3Br
guest molecules in the two types of cages of cubic I
methyl bromide hydrate. Based on previous results the
broad tunnel spectrum is interpreted as a superposition
of four bands, three from inequivalent adsorption sites
in the large cages and one from guest molecules in the
small cages. Methyl rotational potentials with barrier
heights between 4 and 15 meV are derived. Disorder of
hydrogen bonds in the host ice lattice makes the local
potentials vary in bands dV (FWHM) around the
mean values <V> with δV/<V> ~ 50%. The almost continuous distribution of barrier
heights leads to glasslike quasielastic scattering with
little variation of the intensity profiles over a temperature
regime from T = 50 to 150 K. Methyl rotation gets mixed
with whole molecule rotation in the cage above T = 50
K. [ref.
31]
CH3Cl - 5.75H2O - type I. Methyl rotational tunneling, quasielastic spectra and phonon densities of states measured with neutrons at energy transfers from 35 μeV to 40 meV are used to characterize the adsorption sites and potentials of CH3Cl guest molecules in the two kinds of cages of cubic I methyl chloride hydrate. The model of adsorption to hydrogen bonds of the ice cage explains the broad tunnel spectrum as a superposition of four bands. Up to a temperature T ∼ 80 K the quasielastic scattering is influenced by quantum motion. Only above T ∼ 80 K does the effective linewidth follow an Arrhenius behavior with an average activation energy. Rotational potentials contain a significant sixfold component supporting the proposed adsorption to hydrogen bonds with their mirror symmetry. Positional disorder of protons in the hydrogen bonds of the host ice lattice leads to potential distributions of large relative widths δV/V of the order of 0.35. [ref.
34]