Ammonium cyanate: "a two centuries" story
The chemical transformation of ammonium cyanate into urea has been of interest to many generations of scientists, since its discovery by Friedrich Wöhler in 1828. Although widely studied both experimentally and theoretically, several mechanistic aspects of this reaction remain to be understood. Knowledge of fundamental physico-chemical properties of solid ammonium cyanate is a prerequisite for understanding this solid-state chemical transformation. In this issue, two fundamental axes has been investigated as described below.
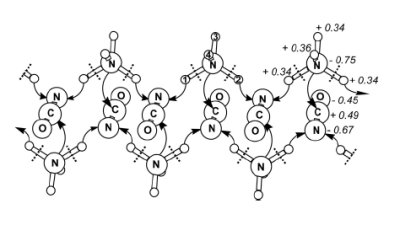
Mechanism of the transformation of crystalline ammonium cyanate into crystalline urea. The atomic charges (in italics) are expressed in a.u. [ref
29]
 Dynamic properties [ref 35]. A comprehensive study of the dynamic properties of the ammonium cation in this material has been performed by means of incoherent quasielastic neutron scattering (QENS) and solid-state 2H NMR spectroscopy. These techniques provide insights into dynamic properties across a complementary range of time scales. The QENS investigations (carried out on a sample with natural isotopic abundances) employed two different spectrometers, allowing different experimental resolutions to be probed. The deuterium NMR experiments (carried out on the deuterated material ND4+.OCN-) involved both NMR line shape analysis and NMR spin-lattice relaxation time measurements. The results of both the QENS and NMR studies demonstrate that the ammonium cation exhibits reorientational dynamics across a wide temperature range, and several dynamic models (based on knowledge of the crystal structure of ammonium cyanate) were considered in this work. It is found that a tetrahedral jump model for the dynamics of the ammonium cation provides the best description for both the QENS and NMR data, and there is excellent agreement between the values of activation parameters established from these two experimental approaches, with estimated activation energies of 22.6 kJ/mol from QENS and 21.9 kJ/mol from NMR spin-lattice relaxation time measurements. The wider implications of the results from this work are discussed in ref 35.
Dynamic properties [ref 35]. A comprehensive study of the dynamic properties of the ammonium cation in this material has been performed by means of incoherent quasielastic neutron scattering (QENS) and solid-state 2H NMR spectroscopy. These techniques provide insights into dynamic properties across a complementary range of time scales. The QENS investigations (carried out on a sample with natural isotopic abundances) employed two different spectrometers, allowing different experimental resolutions to be probed. The deuterium NMR experiments (carried out on the deuterated material ND4+.OCN-) involved both NMR line shape analysis and NMR spin-lattice relaxation time measurements. The results of both the QENS and NMR studies demonstrate that the ammonium cation exhibits reorientational dynamics across a wide temperature range, and several dynamic models (based on knowledge of the crystal structure of ammonium cyanate) were considered in this work. It is found that a tetrahedral jump model for the dynamics of the ammonium cation provides the best description for both the QENS and NMR data, and there is excellent agreement between the values of activation parameters established from these two experimental approaches, with estimated activation energies of 22.6 kJ/mol from QENS and 21.9 kJ/mol from NMR spin-lattice relaxation time measurements. The wider implications of the results from this work are discussed in ref 35.
Exploring the chemical reaction under high pressure [ref
29].In this study, we apply computational methods to investigate the behaviour of ammonium cyanate in the solid state under high pressure, employing a theoretical approach based on the self-consistent-charges density-functional tight-binding method (SCC-DFTB). The ammonium cyanate crystal structure was relaxed under external pressure ranging from 0 to 700 GPa, leading to the identification of five structural phases. Significantly, the phase at highest pressure (above 535GPa) corresponds to the formation of urea molecules. At ca. 25 GPa, there is a phase transition of ammonium cyanate (from tetragonal P4/nmm to monoclinic P21/m) involving a rearrangement of the ammonium cyanate molecules. This transformation is critical for the subsequent transformation to urea. The crystalline phase of urea obtained above 535GPa also has P21/m symmetry (Z = 2). This polymorph of urea has never been reported previously. Comparisons to the known (tetragonal) polymorph of urea found experimentally at ambient pressure suggests that the new polymorph is more stable above ca. 8 GPa. Our computational studies show that the transformation of ammonium cyanate into urea is strongly exothermic (enthalpy change –170 kJ mol–1 per formula unit between 530 and 535 GPa). The proposed mechanism for this transformation involves the transfer of two hydrogen atoms of the ammonium cation towards nitrogen atoms of neighbouring cyanate anions, while the remaining NH2 group creates a C–NH2 bond with the cyanate unit. [ref
29]